Long-term effects of popular acid reflux medications
source : http://www.foxnews.com/health/2013/06/20/long-term-effects-popular-acid-reflux-medications/
source : http://www.foxnews.com/health/2013/06/20/long-term-effects-popular-acid-reflux-medications/

The U.S. Food and Drug Administration on Thursday urged medical device makers and medical facilities to upgrade security protections to protect against potential cyber threats that could compromise the devices or patient privacy. It released that advisory in coordination with a separate alert from the Department of Homeland Security, which disclosed vulnerability in a wide variety of medical equipment that can make those devices vulnerable to remote attacks from hackers. “Over the past year, we've become increasingly aware of cyber security vulnerabilities in incidents that have been reported to us,” William Maisel, deputy director for science at the FDA's Center for Devices and Radiological Health, said in an interview. “Hundreds of medical devices have been affected, involving dozens of manufacturers,” Maisel said, adding that many were infected by malicious software, or malware. But he said all the infections appeared to be unintentional, largely due to malware and computer viruses that were circulating in hospital computer networks and jumped onto the devices. An alert published on the government's Industrial Control Systems Cyber Emergency Response Team website, cited research from Billy Rios and Terry McCorkle of the cyber security firm Cylance Inc, who said they have identified more than 300 pieces of medical equipment that are vulnerable to cyber attack. They include surgical and anesthesia devices, ventilators, drug infusion pumps, patient monitors and external defibrillators. The problem with the equipment is that it can be controlled using default passwords that can be obtained with relative ease by motivated hackers, Rios said in an interview. Those passwords give their holders complete control of the devices and in some cases can be used to gain that access remotely via the Internet, he said. “Somebody could take over the device and make it do whatever they want it to do and it would be almost impossible for hospital staff to know that it had been tampered with,” Rios said. Rios and McCorkle are among a group of security experts who in recent years have suggested that medical devices such as insulin pumps and pacemakers could be vulnerable to hacking. The FDA on Thursday said it is not aware of any patient injuries or deaths associated with devices and hospital computer networks that have been infected with malware and computer viruses. In an advisory on its website, however, the FDA said manufacturers, hospitals and patients need to protect themselves better from the introduction of malware in medical equipment and unauthorized access to settings that control devices. “Many medical devices contain configurable embedded computer systems that can be vulnerable to cybersecurity breaches,” the agency said. The risk of breaches has grown as devices have become increasingly interconnected, via the Internet, hospital networks, other medical devices and smartphones, the FDA said. “Specifically we recommend that manufacturers review their cybersecurity practices and policies to assure that appropriate safeguards are in place to prevent unauthorized access or modification to their medical devices or compromise of the security of the hospital network that may be connected to the device,” the agency said. Among its recommendations, the FDA said manufacturers need to take steps to limit unauthorized device access to trusted users only, particularly for devices that are “life sustaining” or could be directly connected to hospital networks. User IDs, passwords and other security controls need to be strengthened, including potential use of biometrics, the agency said. Moreover, manufacturers need to assure that devices recover and continue to work once security has been compromised. “Cybersecurity incidents are increasingly likely,” the FDA said, “and manufacturers should consider incident response plans that address the possibility of degraded operation and efficient restoration and recovery.” The FDA also urged health care facilities to evaluate their network security, including restricting unauthorized access to the network and networked devices.source : http://www.foxnews.com/health/2013/06/14/fda-urges-protection-medical-devices-from-cyber-threats/
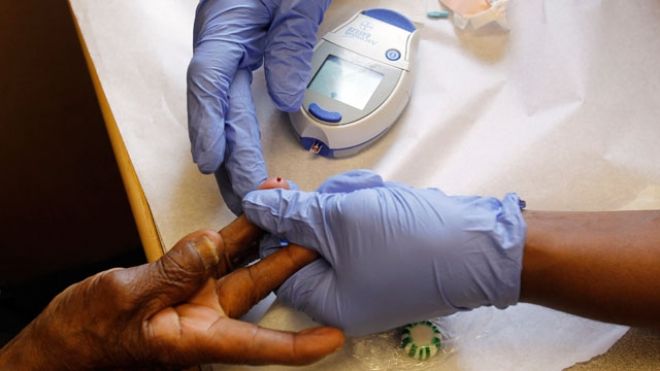
Global drugmakers said on Wednesday they would cooperate with an independent review to address concerns of a potential link between widely used diabetes medicines and pancreatic cancer and other safety problems. The American Diabetes Association (ADA) this week called for a new evaluation of clinical data on drugs used to control blood sugar for patients with type 2 diabetes. They include Merck & Co's $4 billion a year Januvia franchise, Novo Nordisk's Victoza, and Byetta and Onglyza from Bristol-Myers Squibb Co and AstraZeneca Plc, among others. “People who are taking these medications, or who may consider taking them, should have the benefit of all that is currently known about their risks and advantages in order to make the best possible decisions about their treatment,” Dr. Robert Ratner, ADA's chief scientific and medical officer, said in a statement. The medicines are called incretin mimetics because they mimic hormones the body produces to stimulate release of insulin, and are from classes of drugs known as GLP-1 receptor agonists and DPP-4 inhibitors. GLP-1 drugs boost insulin production by the pancreas and slow absorption of food. DPP-4 inhibitors block an enzyme the breaks down the GLP-1 peptide in the gut, thereby increasing insulin production. The U.S. Food and Drug Administration in March said it was studying unconfirmed reports that the drugs cause inflammation of the pancreas and pre-cancerous changes to cells in the pancreas. European health regulators are also studying the issue. Around the same time, new concerns arose from a small study conducted by a leading diabetes expert, Dr. Peter Butler, from the University of California, Los Angeles. Butler examined human pancreases from patients who had died of causes unrelated to pancreatic disease and found more pancreas lesions and one cancerous tumor in those who had taken Januvia or Byetta compared with nondiabetics or diabetics who had not taken those medicines. More than 370 million people are living with diabetes worldwide, with type 2 accounting for 90 percent to 95 percent of the cases, according to the International Diabetes Federation. Without significant lifestyle changes to curb obesity and other causes of diabetes, that number could balloon to as many as 552 million by 2030, the organization projected. NO FIRM EVIDENCE OF RISK Bristol-Myers and AstraZeneca issued a joint statement expressing support for the ADA initiative. Merck said it is committed to participating in the ADA effort, and is separately conducting a 14,000-patient heart safety study of Januvia. Danish drugmaker Novo Nordisk, in an emailed comment, said its studies to assess safety signals of its $1.8 billion a year Victoza do not reveal any evidence of increased risk of pancreatitis or pancreatic cancer. “As far as the ADA proposal is concerned, this is something that I do support from a conceptual basis,” said Alan Moses, Novo's global chief medical officer, in a telephone interview. “In principle, Novo Nordisk absolutely supports working with the other companies, but the final details depend on what specifically is being proposed,” Moses said, adding “it all depends on the credibility of the data that's being evaluated.” The safety concerns are being discussed on Wednesday and Thursday at a workshop conducted by the National Institutes of Health (NIH) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). A document outlining the FDA's presentation at the workshop was handed out at the meeting. Attendees quoted it as saying the agency review of trial data on the drugs did not provide enough evidence to say whether there is a link to pancreatitis, and that long-term study would be required to determine any cancer risk. A FDA spokeswoman could not immediately confirm the contents of the document. “The FDA basically appears to just be saying 'look, there just isn't enough information here to make an informed decision',” said Mark Schoenebaum, a drug industry analyst with ISI Group who was attending to two-day NIH/NIDDK workshop. “There are no conclusions at this point in time from the meeting and I doubt very much there will be because the science quite frankly is immature about the whole area of pancreatic cancer,” said Novo's Moses, who will address the workshop on Thursday. “Everybody agrees that this is an incredibly complex area.” In an interview with Reuters earlier this week Peter Stein, Merck vice president of clinical research for diabetes, expressed full confidence in the safety of Januvia. During the workshop, Merck said it will present data on the safety profile of Januvia, known chemically as sitagliptin, including an updated analysis of data from more than 14,000 patients from 25 randomized clinical trials. Merck last month reported a surprising decline in first quarter sales of Januvia, which has been the drugmaker's fastest growing medicine since its 2006 approval. It was not clear if increased competition, safety concerns or other factors led to the 4 percent slip in quarterly sales to $884 million.source : http://www.foxnews.com/health/2013/06/13/drugmakers-to-cooperate-in-safety-review-diabetes-drugs/

U.S. health advisers voted on Thursday to recommend relaxing market restrictions on GlaxoSmithKline's diabetes drug Avandia, the former blockbuster at the center of one of the biggest drug controversies in recent years. The vote, by a divided Food and Drug Administration advisory committee of outside health experts, could modestly enlarge the market for Avandia in the United States and lay the groundwork for further research into the drug's health risks. FDA will now take the vote into consideration for a final decision on how the pill also known by the generic name rosiglitazone can be used. The committee did not consider a specific change in protocol. But 13 experts on the 26-member panel who backed modification said current restrictions that require prescribing physicians and pharmacists to be certified should be relaxed or eliminated after a reexamination of Glaxo safety data settled longstanding concerns about the danger of death from cardiovascular disease. “In general, this drug doesn't look any different than any other diabetes drug,” said Dr. William Hiatt, a cardiologist from the University of Colorado, who was among seven experts who backed lifting restrictions altogether. Five committee members favored keeping the current sales restrictions, while one voted to withdraw Avandia from the market altogether. Glaxo, which no longer plans to promote Avandia, issued a statement saying the company would work with FDA as it considers its decision. “We continue to believe that Avandia is a safe and effective treatment option for type 2 diabetes when used for the appropriate patient and in accordance with labeling,” Dr. James Shannon, Glaxo's chief medical officer, said in a statement. The British drugmaker's stock closed nearly 1.5 percent lower in London trading before the committee voted. Avandia was once the world's best-selling treatment for type 2 diabetes, with annual sales of $3.2 billion. In 2010 its use in the United States was heavily restricted and it was withdrawn from the market in Europe because of the possibility of increased risk of heart attack and stroke. Only 3,000 people in the United States take it today, down from about 120,000 just before the restrictions were put in place. Much of the advisory committee's two-day meeting focused on a Duke University reexamination of a Glaxo safety study known as Record that confirmed initial findings of no significant increased heart risk from the drug. That reassured some experts that earlier concerns with the quality of the research had been unwarranted and encouraged support for the restrictive protocols, while retaining continued guidance on potential dangers for patients and care providers. A departing train But others said the original data was incomplete and compiled through a flawed study design, while other research pointed to the possibility of significant increased risk of cardiovascular death. “When you look at the overall totality of evidence, it is not sufficient enough to either implicate or exonerate rosiglitazone versus cardiovascular risk,” said advisory panel member Dr. Sanjay Kaul of the Cedars-Sinai Heart Institute. Several committee members endorsed suggestions for a major new clinical trial to determine precisely the drug's risks in the face of a growing worldwide diabetes threat. But other experts concluded that years of negative publicity made funding major research unfeasible except for new diabetes treatments now in the pharmaceutical pipeline. “The train has left the station,” said Gerald van Belle, director of the Clinical Trials Center at the University of Washington. Experts including the advisory committee's chairman, Dr. Kenneth Burman of the Washington Hospital Center, favored the creation of a registry to monitor the health of patients who currently take the drug if major studies into safety and efficacy were not an option. Some experts view Avandia as a potential alternative to other diabetes treatments, including insulin, that could become more important as the incidence of obesity and diabetes grows, bringing with it a host of costly chronic ailments ranging from heart and kidney disease to blindness and dementia. “When treating diabetes we really do need drugs that lower blood sugar without causing hypoglycemia, and there's not a lot that's available,” Dr. Ellen Seely of Harvard Medical School. “When you're dealing with individual patients, you come up often against dead ends on what you can do. And it's important to have options,” she said. But panel members agreed the market potential for Avandia may never again be large. Glaxo has settled lawsuits filed by tens of thousands of U.S. patients who had taken Avandia and claimed Glaxo failed to inform them about safety risks. Several thousand other cases remain pending. The drugmaker last July agreed to pay $3 billion to settle what U.S. officials called the largest case of healthcare fraud in U.S. history. The agreement resolved allegations that Glaxo failed through 2007 to provide the FDA safety data on Avandia and that the company improperly marketed other drugs.source : http://www.foxnews.com/health/2013/06/07/fda-panel-votes-to-relax-avandia-restrictions/
I am now totally convinced that our current federal government loves confusion. When you have a single agenda, and many ways to spin it, the American public never gets a clear answer and that is exactly what has happened with the Plan B emergency contraception controversy. A U.S. appeals court ruled on Wednesday that the U.S. Food and Drug Administration (FDA) must make certain forms of the emergency contraception pill available to children of all ages, without a prescription. This is exactly what I have been warning the American public about. One has to remember that the FDA first approved this form of over-the-counter contraception for women of all ages back in 2011. When that initial FDA ruling came out, there was a loud public outcry and restrictions were quickly put in place barring women under the age of 17 from purchasing these pills. But of course, that was just one spin on the story. In April, a New York judge ruled that restricting access to Plan B was inappropriate, forcing the FDA to reconsider their initial finding that emergency contraception should be available to children of all ages. And then, we got a third spin on the story, as the FDA tried to lower the age limit for access to emergency contraception to15 in May. There was another outcry and more criticism, because we know perfectly well that a 15-year-old may not have a clear understanding of how to utilize emergency contraception. Now, we see that an appeals court is forcing the FDA to do what they wanted to do in the first place. How convenient. And the final ruling is still unclear, after the court decided on Wednesday that while the two-pill version of emergency contraception can now be sold over-the-counter to women of all ages, the one-pill version will still only be sold to women age 17 or older. The court did not explain its reasoning. While there is still a lot of confusion about the ruling, it seems as though the FDA will ultimately get its way. So, what’s the message here?

I am now totally convinced that our current federal government loves confusion. When you have a single agenda, and many ways to spin it, the American public never gets a clear answer and that is exactly what has happened with the Plan B emergency contraception controversy. A U.S. appeals court ruled on Wednesday that the U.S. Food and Drug Administration (FDA) must make certain forms of the emergency contraception pill available to children of all ages, without a prescription. This is exactly what I have been warning the American public about. One has to remember that the FDA first approved this form of over-the-counter contraception for women of all ages back in 2011. When that initial FDA ruling came out, there was a loud public outcry and restrictions were quickly put in place barring women under the age of 17 from purchasing these pills. But of course, that was just one spin on the story. In April, a New York judge ruled that restricting access to Plan B was inappropriate, forcing the FDA to reconsider their initial finding that emergency contraception should be available to children of all ages. And then, we got a third spin on the story, as the FDA tried to lower the age limit for access to emergency contraception to15 in May. There was another outcry and more criticism, because we know perfectly well that a 15-year-old may not have a clear understanding of how to utilize emergency contraception. Now, we see that an appeals court is forcing the FDA to do what they wanted to do in the first place. How convenient. And the final ruling is still unclear, after the court decided on Wednesday that while the two-pill version of emergency contraception can now be sold over-the-counter to women of all ages, the one-pill version will still only be sold to women age 17 or older. The court did not explain its reasoning. While there is still a lot of confusion about the ruling, it seems as though the FDA will ultimately get its way. So, what’s the message here? The polarizing health care agenda of this federal government is like a train without a stop and parents need to be aware of this. As I have said before, this is a medication. Yes, I know that it is safe – but it does have side effects. Side effects to Plan B can include, but are not limited to: migraines, high cholesterol, high blood pressure and blood clots. If this drug is available to young children, it might lead to problems including the misuse of the medication and the risk that children will utilize this as a regular form of contraception. Furthermore, it will exclude parents from the decision-making process. And in my opinion, parents can be very valuable in counseling children about proper behavior and doing the right thing. This is taking parents and caregivers out of the equation and inhibiting their ability to help their children live a healthy and happy life. America, we are being bamboozled. Wake up and pay attention.source : http://www.foxnews.com/health/2013/06/05/dr-manny-government-must-stop-bamboozling-americans-about-plan-b/

The top-selling class of blood-pressure drugs is under attack from an unusual source: a senior regulator at the Food and Drug Administration. Bucking his bosses, Thomas A. Marciniak is seeking stronger warnings about the drugs known as angiotensin receptor blockers, or ARBs, according to internal documents reviewed by The Wall Street Journal. The drugs, which are taken by millions of people and generated $7.6 billion in U.S. sales in 2012, may be linked to higher cancer rates, Dr. Marciniak argues, a view shared by some outside doctors. Top FDA officials say evidence doesn't support a link. The debate over ARBs highlights the question of whether the U.S. drug-safety agency devotes enough effort to examining the safety of long-marketed blockbusters as it focuses on new drugs. In a rare rebellion by an FDA reviewer, Dr. Marciniak has clashed with his bosses over his desire to spend time on ARB safety, instead of just on new-drug applications. Ellis Unger, chief of the drug-evaluation division that includes Dr. Marciniak, called the complaints a “diversion,” and said in an interview, “We have no reason to tell the public anything new.” ARBs on the market in the U.S. include Novartis AG's Diovan, Daiichi Sankyo Co.'s Benicar, Merck & Co.'s Cozaar, Boehringer Ingelheim GmbH's Micardis; Avapro, from Sanofi SA and Bristol-Myers Squibb Co.; and AstraZeneca PLC's Atacand. Patients take these medicines daily to avoid heart attack, stroke and heart failure. In a 2010 study published in Lancet Oncology, Ilke Sipahi and colleagues at University Hospitals in Cleveland looked at five studies involving 68,402 patients and found that people taking ARBs had an 11 percent greater risk of new cancer overall and a 25 percent greater risk of new lung cancer, compared with patients who didn't get the drugs. But within a year, the FDA gave the all-clear signal, saying its own analysis found “no increase in risk” from taking ARBs. Europe's drug regulator also dismissed the cancer concerns. Dr. Marciniak wasn't persuaded. In its analysis, the FDA combined different studies to look at more patients, multiplying its statistical power to find possible side effects from the drugs, a technique called meta-analysis. If the original studies have flaws, however, meta-analyses can simply compound the problem, researchers say. That is what happened to the FDA, says Dr. Marciniak, who warned others in the agency that taking results tabulated by companies was likely to produce unreliable results: “Garbage in, garbage out,” he wrote. Among other things, Dr. Marciniak said in an internal analysis viewed by the Journal that the FDA meta-analysis didn't count cases of “lung carcinomas” as lung cancers, which they are. Click for more from The Wall Street Journal.source : http://www.foxnews.com/health/2013/05/31/dispute-flares-within-fda-over-safety-popular-blood-pressure-drugs/

Allergy medications may help you get through the spring and summer months, but it's important to know that the drugs could affect your ability to drive, the Food and Drug Administration is reminding consumers. These medications, which contain antihistamines, can sometimes cause drowsiness and slower reaction times, the FDA said. Consumers should read the drug facts label on their medication to see whether drowsiness is a side effect. If an allergy medication causes drowsiness, people need to be cautious about deciding to drive or operate machinery, the FDA says. People should avoid using alcohol, sedatives (sleep medications) and tranquilizers when taking allergy medication because these substances may increase drowsiness. [See Will Allergies Be Worse in 2013?] Those who switch to a new antihistamine drug should not assume they can take the same dose as they did with the older drug, the FDA says. Different allergy medications may be dosed differently, and people may need to alter the dose they take. People should not take more than the recommended dose. If the correct dosage isnt providing you the relief you expect, dont simply keep taking more and more of that product, FDA pharmacist Ayana Rowley said in a statement. Instead, people should consult a health care professional, Rowley said. Allergy sufferers should be aware that some allergy medications take longer to work than others. In addition, the drowsiness you feel after taking the medication may last some time, including into the next day, the FDA said. Copyright 2013 LiveScience, a TechMediaNetwork company. All rights reserved. This material may not be published, broadcast, rewritten or redistributed.source : http://www.foxnews.com/health/2013/05/29/allergy-meds-can-pose-driving-hazard-fda-says/
In the summer of 2011, Andrea Syglowski noticed a mole she’d had her entire life was starting to look different. Concerned, she booked an appointment with a dermatologist, and within a week was diagnosed with stage-0 melanoma – an early phase of the deadliest type of skin cancer. A week later, Syglowski, a Philadelphia-based actress who is in her 20s, underwent surgery to remove the mole, leaving her with a five-inch scar on her leg. Syglowski said that while she used sunscreen before her diagnosis, she now realizes she didn’t always use it correctly. “I think all I knew was that I needed to have it on,” Syglowski told FoxNews.com. Skin cancer affects millions of people like Syglowski every year, but many remain confused about the basic rules of sunscreen application. However, the Food and Drug Administration (FDA) has recently implemented new changes to sunscreen labeling, which aim to clear up some of the confusion. Here’s what to look for on sunscreen labels this summer. ‘Broad spectrum’ protection There are two types of ultraviolet light: UVA and UVB rays.  Currently, all sunscreens contain UVB protection, which shields the skin against cancer-causing sunburns. But not all sunscreens are required to have UVA protection, which protects against both skin cancer and aging. “UVB is what causes a sun burn; UVA doesn’t sunburn you. But now, we want something more. We want sunscreens to prevent cancer and wrinkles in addition to sunburn,” Dr. James Spencer, a member of the American Academy of Dermatology and a board certified dermatologist in St. Petersburg, Fla., told FoxNews.com. According to the new FDA guidelines, new sunscreen labels can only claim that they offer “broad spectrum protection” if they protect against both UVA and UVB rays. “When we say broad spectrum, we mean we're covering both UVA and UVB as wide in the spectrum as we can cover,” pharmacist Ian Ginsberg, owner of C.O. Bigelow in New York City, told FoxNews.com. Furthermore, sunscreens can now only claim to “prevent cancer” and “prevent wrinkles” if they contain both UVA and UVB protection. “It turns out that UVA contributes to cancer and wrinkles but not to burns. So now we’re asking sunscreens to help (protect us) from cancer and wrinkles too; we want that UVA added in,” Spencer said. ‘Water resistant’ not ‘waterproof’ Waterproof sunscreen may sound like a great option, but according to Spencer, “there’s no such thing as waterproof.” Sunscreen companies must now remove the word “waterproof” from their labels and replace it with the phrase “water resistant,” according to the new FDA guidelines. A “water resistant” sunscreen will be less likely to wash off in water, but doctors warn that it still needs to be reapplied. To make sure people remember to lotion up again after getting wet, the FDA now requires all sunscreens to state whether they are water resistant for “40 minutes” or “80 minutes.” “So that gives you an idea…how long you’re good for; that’s useful information,” Spencer said. And if you’ve had the same tube of sunscreen for the entire summer – you’re doing something wrong. “One tube should only last two weeks, and if you’re going to the beach, (it should last) for a week,” Dr. Hooman Khorosani, assistant professor of dermatology and chief of division of Mohs, reconstructive and cosmetic surgery at the Icahn School of Medicine at Mount Sinai in New York City, who also treated Syglowski, told FoxNews.com. Don't forget to take note of the expiration date on your tube of SPF either. Expired sunscreen could be less effective.  “Most people throw these things in their beach bag and/or let them sit in direct sunlight for hours on end so you should live by the (expiration) date,” Ginsberg said.  SPF 30, not SPF 100 Resist the urge to reach for the highest SPF on the shelf.  It likely won’t offer any more protection, according to doctors. “An SPF 30 blocks 97 percent of UBV rays - SPF 45 blocks 98 percent. Once you get to 98 percent, it’s getting a little silly. And SPF 100 is a little misleading,” Spencer said. “You can’t get more than 100 percent blocked.” Spencer said the FDA is considering prohibiting sunscreens labeled higher than SPF 50, but due to objections from sunscreen companies, the change is still being negotiated.   In the meantime, Spencer recommends looking for an SPF of at least 30 and reapplying sunscreen every few hours. Syglowski – who has now been melanoma-free for nearly two years – says she is much more vigilant about sunscreen application and schedules regular skin checks with Khorosani – something she encourages other women to do as well. As for Khorosani, he said one technique seems to be particularly effective at encouraging patients to be vigilant about sun protection. “I have a photo of a patient who sat by the window every day. The left side of her face, which was facing the window, was being hit by (wrinkle-causing) UVA rays, so it looks like she’s 50. The right side looks like she’s 35,” Khorosani said. “All I need to do is show that photo to the women who come in.”  source : http://www.foxnews.com/health/2013/05/22/heading-to-beach-new-spf-regulations-issued-by-fda/

WASHINGTON – & Sunbathers headed to the beach this summer will find new sunscreen labels on store shelves that are designed to make the products more effective and easier to use. But despite those long-awaited changes, many sunscreens continue to carry SPF ratings that some experts consider misleading and potentially dangerous, according to a consumer watchdog group. A survey of 1,400 sunscreen products by the Environmental Working Group finds that most products meet new federal requirements put in place last December. The rules from the Food and Drug Administration ban terms like “waterproof,” which regulators consider misleading, and require that sunscreens filter out both ultraviolet A and B rays. Previously some products only blocked UVB rays, which cause most sunburn, while providing little protection against UVA rays that pose the greatest risk of skin cancer and wrinkles. Despite that broader protection, one in seven products reviewed by the watchdog group boasted sun protection factor, or SPF, ratings above 50, which have long been viewed with skepticism by experts. In part, that's because SPF numbers like 100 or 150 can give users a false sense of security, leading them to stay in the sun long after the lotion has stopped protecting their skin. Many consumers assume that SPF 100 is twice as effective as SPF 50, but dermatologists say the difference between the two is actually negligible. Where an SPF 50 product might protect against 97 percent of sunburn-causing rays, an SPF 100 product might block 98.5 percent of those rays. “The high SPF numbers are just a gimmick,” says Marianne Berwick, professor of epidemiology at the University of New Mexico. “Most people really don't need more than an SPF 30 and they should reapply it every couple of hours.” Berwick says sunscreen should be used in combination with hats, clothing and shade, which provide better protection against ultraviolet radiation. Some dermatologists say there may be some rationale for using higher SPF sunscreens, since users often don't apply enough of the lotion to get its full effect. “The challenge is that beyond 50 the increase in UV protection is relatively small,” says Dr. Henry Lim, chair of dermatology at the Henry Ford Hospital in Detroit. The SPF number indicates the amount of sun exposure needed to cause sunburn on sunscreen-protected skin compared with unprotected skin. For example, a SPF rating of 30 means it would take the person 30 times longer to burn wearing sunscreen than with exposed skin. There is a popular misconception that the SPF figure relates to a certain number of hours spent in the sun. However this is incorrect, since the level of exposure varies by geography, time of day and skin complexion. The FDA itself said in 2011 that “labeling a product with a specific SPF value higher than 50 would be misleading to the consumer.” At the time the agency proposed capping all SPF values at 50 because “there is not sufficient data to show that products with SPF values higher than 50 provide greater protection for users.” But regulators have faced pushback from companies, including Johnson & Johnson, which argue that higher SPF products provide measurable benefits. As a result, the FDA says it is still reviewing studies and comments submitted by outside parties, and there is no deadline for the agency to finalize an SPF cap. It took the agency decades to put in place last year's sunscreen changes. FDA first announced its intent to draft sunscreen rules in 1978 and published them in 1999. The agency then delayed finalizing the regulations for years until it could address concerns from both industry and consumers. The FDA is also reviewing the safety of effectiveness of spray-on products, which use different formulations from other sun-protection solutions. Among other concerns, the agency is looking at whether the sprays can be harmful when inhaled. The survey by the Environmental Working Group found that one in four sunscreens sold in the U.S. is a spray product. “People like the sprays because they are quick to put on and cover a lot of area,” said Dr. Darrell Rigel, a dermatologist in New York.  ”The downside is that you usually have to apply two coats.” More than 76,000 men and women in the U.S. will be diagnosed with melanoma this year and 9,480 are expected to die from the aggressive form of skin cancer, according to the National Cancer Institute. The disease, which is often linked to ultraviolet exposure, is usually curable when detected early.source : http://www.foxnews.com/health/2013/05/20/consumer-group-flags-high-spf-ratings-on-sunscreenas-misleading/